Large-scale Extraction and Purification of Enzymes and other Proteins
In the recent surge of interest in biotechnology considerable emphasis has been placed upon advances in the genetic aspects as well as in fermentation technology. Biotechnologists have however, always appreciated that the techniques for extraction and purification of proteins from an inseparable part of the whole biotechnological process. There has, in the last few years, been an increasing need to ally protein purification techniques to large-scale fermentation. Sometimes on a vast scale involving thousands of litres of products, to purify a range of proteins from microbial cultures. Proteins which are to be administered to patients pose special problems. These cannot adequately be covered in 2 chapter devoted to the wider problem of purification, but in general, very high standards of hygiene and process control are required. In addition, the use of techniques or materials which could result in any contamination of the final product must be carefully evaluated in terms of product safety and acceptability to the licensing authorities.
This chapter will deal mainly with the isolation and purification of proteins from microbial sources, but the techniques described can and do apply equally well to the extraction of materials from plant and animal tissue.
Bacterial protein can be conveniently classified into extracellular bound and free intracellular proteins. Although the purification of intracellular proteins necessitates some cell breaking process, once this has been achieved then, except that the volumes involved may be smaller than for extracellular proteins, there is no fundamental difference in the purification procedures for those two groups of proteins.
Before describing in detail the individual methods involved in isolation and purification of proteins, it is worth considering the relationship between small-scale and large-scale processes. There are many techniques available, but not all are suitable for large-scale work. This may result from some inherent restriction in the available technique or apparatus or from the increase in time which usually accompanies the transition to a large-scale process. Effective bacterial disruption for example, can be accomplished by sonification, freezing and thawing, grinding or solid shear. The scale-up of these techniques in many cases is not practical, and in others the result is inadequate. Similarly, the use of ultracentrifugation is a valuable laboratory technique which cannot be conveniently used for large-scale processes.
The definition of large-scale is a matter of opinion, but in the context of this chapter will refer to processes carried out with one or more kilograms of wet cell paste. The term 'extraction' will be used to define the methods by which microorgansims are disrupted, and the terms 'isolation' and 'purification' will relate to the other techniques involved in obtaining a pure, or specified, product.
EXTRACTION BY CHEMICAL METHODS
Alkali
This method has been used with considerable success in both small- and large-scale extractions of various bacteria. Wade isolated the therapeutic enzyme L-asparaginase (3,5,11-L-asparagine amido-hydrolase) by exposing bacteria to an alkaline pH between 11 and 12.5 for 20 minutes. Salton has reported the use of alkali treatment in the preparation of plant, fungal and bacterial cell walls, and the degree of hydrolysis was fairly extensive as membrane-bound components such as cytochrome-C were also released. The success of alkali treatment is dependent on the stability of the required enzyme to high pH. It has also been suggested that this method may inactivate proteases should they be present, and reduce the possibility of pyrogen contamination of therapeutic enzyme preparations. Such methods have possible applications for rapid inactivation and lysis of genetically engineered mocro-organisms.
Lysozyme and EDTA
Lysozyme is an enzyme, produced commercially from hen egg white, which specifically catalyses the hydrolysis of b-1-4-glycosidic bonds in the mucopeptide moeity of bacterial cell walls. Gram-positive bacteria, which rely to a greater extent on the wall mucopeptide for rigidity than Gram-negative bacteria, are more susceptible to lysozyme than Gram-negative bacteria. However, final rupture of the cell envelope often depends upon the osmotic pressure of the suspending medium once the cell wall has been disrupted. In Gram-negative bacteria the breakdown of the cell wall is rarely achieved by lysozyme alone. The addition of EDTA, which has been shown to cause release of lipopoly-saccharide from Gram-negative cell envelopes is a necessary adjunct. It has been suggested that EDTA acts by chelation of divalent cations essential for wall stability, and that this then allows access of lysozyme to act on the mucopeptide layer.
This technique is rarely used for the large-scale extraction of bacterial enzymes because of the relatively high cost of lysozyme, although this is small in comparison with the value of a purified enzyme. Crude hen egg white is much cheaper and often equally effective, and experiments have been carried out with immobilized lysozyme. Lysozyme is a very gentle method of cell lysis and is to be favoured if the enzyme is susceptible to the temperature rise during physical lysis procedures. It has recently been used for the lysis of a Pseudomonas fluorescents prior to the purification of an aryl acyl hydrolase.
Detergents
Detergents are either ionic, for example sodium lauryl sulphate (anionic) and cetyldiethylammonium bromide (cationic) or non-ionic, for example Tweens and Tritons. Under conditions of low ionic strength and appropriate pH, detergents will combine with lipoprotein to from micelles. Therefore, the lipoprotein constituents of biological membranes can be solubilized, or the membranes made permeable. The mechanism of detergent/lipoprotein complex formation is still not fully understood, but is thought to involve electrostatic (salt links) and Vander Waals' forces. These complex formations appear to be critically dependent upon pH and temperature.
Ionic detergents are more reactive than non-ionic detergents and can lead to the dissociation of lipoproteins. This in turn can further lead to protein denaturation, precipitation, and occasionally, to the hydrolysis of peptide bonds. For this reason, detergents are not desirable in the extraction of enzymes. Further, salt precipitation of proteins is made difficult in the presence of detergents. However, this can be overcome in many cases by the use of ion exchange chromatography.
Nevertheless, detergents do have considerable use in some extraction processes. Morris indicated their usefulness in the fractionation of virus particles. Triton X-100 was used for the large-scale release of cholesterol oxidase from Nocardia sp. and sodium cholate was used to solubilize pullulanase (pullulan-6-glucan hydrolase), a membrane-bound enzyme from intact cells of Klebsiella pneumoniae.
Cold Shock
The effect of cold shock (a rapid reduction from normal growth temperature to 0°C) was first observed by Sherman. The literature on this phenomenon indicates that Gram-negative bacteria are more susceptible than Gram-positive organisms. The effect which only occurs under specialized conditions, results in a loss of viability and release into the surrounding medium of 260 nm adsorbing material, amino acids, and ATP.
However, as a technique for bacterial disruption on a large-scale, cold shock would be difficult to employ. Two of the major limitations imposed by this technique are firstly that cold shock has little or no effect on cell suspensions with a density greater than 108/ml and secondly bacteria are considerably more susceptible to cold shock when in the exponential growth phase. On the large scale the size of vessel and sophistication of equipment required to ensure success with this method would be unpractical.
Osmotic Shock
Osmotic shock has been used in the extraction of hydrolytic enzymes and binding proteins from a number of Gram-negative bacteria, including Salmonella typhi-murium and E. coli, and in the release of DNA from T (even) bacteriophages by rupture of the protein coat. The method involves washing the bacteria in buffer solution to free them from growth medium and then resuspending them in 20% buffered sucrose. The cells are then allowed to equilibrate, resulting in the loss of some internal water and are then removed from suspension by centrifugation. The cell paste obtained is rapidly dispersed in water at approximately 4°C. It has been suggested that the sudden increase in the osmotic pressure inside the cell causes the release of certain cell constituents but it is clear that this method is in some respects similar to that of cold shock (see above).
Only 4-7% of the total bacterial protein is released by osmotic shock, although in many cases viability is drastically reduced. If the required enzyme is located in the periplasmic region, osmotic shock and cold shock are both worth considering as a 14- to 20-fold increase in purification may be obtainable compared to other disruptive techniques. Osmotic shock has proved to be particularly useful for the extraction of periplasmic aminoglycoside inactivating enzyme, for example kanamicin acetyl transferase from E. coli.
It is also a valuable technique from the release of enzymes from marine bacteria, such as luciferase from Photobacterium fischeri. In the case of marine organisms it is only necessary to resuspend the cells in dilute buffer to achieve lysis as the growth medium contains 30 g/1 of NaCl. Osmotic shock is not suitable for enzyme release from Gram-positive bacteria and in part, this can be explained by the fact that several Gram-positive cocci have internal osmotic pressures in the region of 20 atmosphered. The integrity of the cell is maintained by a rigid mucopeptide layer in Gram-positive bacteria, and this may enable them to maintain such internal osmotic pressures, whereas Gram-negative organisms cannot.
Extraction by Physical Methods
Sonication
The term ultrasonics is used to denote the frequencies above the range of human hearing, such frequencies are 20 kHz and above. When expressed in wavelength, ultrasonic waves in liquid range from 6 to 2.4 X 10-4 cm. This brings them into a similar order of magnitude to those of visible light.
The application of ultrasonics in liquid creates the phenomenon known as cavitation. Areas of compression and rarefaction occur, and cavities, which form in the areas of rarefaction, rapidly collapse as the area changes to one of compression. The bubbles produced in the cavities are thus compressed to several thousand atmsopheres. Subsequent to their collapse, shock waves are formed, and these shock waves are considered to be the destructive element in this procedure.
Ultrasonic oscillators have been successfully used in a number of extraction procedures. Salton have employed this method for the preparation of bacterial cell walls. The effect on protozoa, Infusoria, and common water weed, Elodea, is one of non-selective destruction.
Others have reviewed this method of cell disintegration in relation to enzyme release. However, it should be noted that some bacteria, for example Staphylococci are very resistant to ultrasonic disruption. In addition, membrane-bound proteins, such as those related to electron transport systems may require considerable exposure to achieve solubilization.
The efficiency of ultrasonic treatment depends upon various environmental parameters including pH, temperature, and ionic strength of the suspending medium as well as the time of exposure. The selection of a set of conditions is essentially an empirical choice, and the conditions will vary with the particular organism as well as the required product.
Although ultrasonic treatment has proved to be a useful and versatile method for laboratory-scale work, its application to the disruption of quantities of bacteria is limited by the difficulty of transmitting sufficient power to large volumes of suspension.
Freezing and Thawing
The effects of freezing and thawing on micro-organisms are in some ways similar to those observed during cold and osmotic shock. However, in addition to rapid cooling and intracellular and extracellular solute concentration, the formation of intracellular and extracellular ice crystals result in further damage to the cell. Mazur reviewed the various physicochemical events leading to injury by freezing. The efficiency of freezing and thawing for general protein release is however, limited, and less than 10% of the total soluble protein is normally liberated, even from Gram-negative bacteria by a single operation. Proteins located in the periplasmic space can be released more readily, however, and up to 60% of an R-factor mediated Penicillinase from E. coli appeared in the supernatant after the cells had been frozen and thawed once (Melling, unpublished results). Freezing and thawing is of ten, perhaps unintentionally, an adjunct to other cell-breaking methods since bacterial pastes are often stored at -20°C prior to enzyme extraction and purification.
Freezing and thawing may result in a loss of enzyme activity, have demonstrated reduced protease activity in various strains of Streptococcus lactis after freezing and thawing and subsequent storage at 30°C for ten days. Speck demonstrated that this inactivation is related to changing enzyme structure. On storage at 22°C, after freezing and thawing, the membrane associated protease was found to be in the active monomer and dimer form, but after storage for one day at 30°C the enzyme aggregated to form inactive high molecular weight units. Hanafusa has demonstrated denaturation and inactivation of AT-Pase. Catalase, on the other hand, showed no apparent change in structure but considerable loss of activity. These results indicate that considerable problems may be encountered in using this method for the extraction of enzymes. Furthermore, unless bacterial pastes are frozen in small batches, thawing and subsequent resuspension present problems.
Solid Shear
Buchner was the first to use this method for breaking micro-organisms to release enzymes. They mixed yeast paste and kieselguhr and subjected the mix to hydraulic pressure. The kieselguhr acted as an abrasive, and following application of pressure, zymase was released. A review of these early studies can be found in Alcoholic fermentations.
Hughes developed the fly press. In this process bacterial paste was mixed with an abrasive, or frozen to -20°C in which case the ice crystals formed acted as an abrasive. The paste was placed in a cylindrical hole in a metal block and a tight-fitting metal plunger was then inserted. A pressure of some 10-15 tons per square inch (150-230 MPa) was applied to the top of this plunger. A very high percentage of the cells in a particulate suspension have been found to be ruptured using this method successfully obtained enzyme preparations from bacterial endospores by alternating the Hughes Press with freezing and thawing. Eaton modified the metal block such that it could be separated into two halves, and this facilitated almost complete recovery. However, even using a block with 2 and 3 plungers little more than 30 g of cell paste can be handled, and therefore large-scale work using this method is impossible.
The X-press was developed which is capable of processing 100 g of Saccharomyces cerevisiae per minute. Frozen cells are disrupted by passage through a perforated disc, the outlet temperature being approximately -22°C. Rupture of the cells is thought to be caused by the shear forces exerted by the passage of the extruded paste through the small orifice, the shear being aided by crystal formation in the frozen paste. Solid shear cell disruption has proved to be an excellent method for obtaining enzyme protein and bacterial cell wall preparations.
A larger version of the solid-shear disintegrator has been established by Magnusson which is capable of disrupting up to 10 kg yeast paste per hour. Such equipment is perhaps over complicated for routine use, although the fact that cell breakage occurs at -20°C may be an advantage when heat labile enzymes are being extracted.
Grinding or Agitation with Abrasives
Booth developed a grinding mill for the discaption of bacteria. This did not prove satisfactory as both E. coli and B. subtilis required 2 hours' treatment, and Sarcina leatea-4 hours for enzyme release.
Curran studied the disintegration of bacteria by agitation in the presence of glass beads. King showed the optimum conditions to be (a) beads of 0.13-0.26 mm diameter, (b) 300-500 strokes per minute, and (c) an amplitude of 6 cm.
Mickle developed an apparatus which has found wide application in the disruption of micro-organisms. Its most popular use is in the preparation of microbial cell walls. Walls have been obtained from bacteria, bacterial spores, mycobacteria, and fungi. The apparatus comprises a glass cup with indentations suspended at the end of a variable length armd into the glass cup is placed 10 ml of bacterial suspension (10-20 mg of protein/ml) together with an equal volume of Ballatini beads (0.1-0.2 mm in diameter). The cups are then shaken at a frequency of 50 cycles/sec. Overheating can be avoided by working in a cold room; while the required degree of disruption has to be worked out on an empirical basis. Shockman described a method similar in principle to that of Mickle which could cope with up to 6 g of material per run.
Although the above methods are only suitable for small quantities of bacteria, the same principle has been used for disruption of larger amounts. Rehacek described an apparatus which employed intense stirring of cell suspensions with glass beads. Rehacek reported a modification to the original incorporating a bead separation system and enabling continuous operation to be used with a flow of bacterial suspension through a chamber filled with glass beads.
The disruptive effect was suggested to result from shear forces produced between velocity gradients; such gradients being due to the stream lines. In addition, collisions between organisms and beads occur as well as grinding between rolling beads with different speeds. The degree of disintegration has been found to depend upon the rate of stirring, the concentration of micro-organisms, the concentration and size of beads, and the contact time.
A commercially produced apparatus employing these principles has produced a most efficient method for the disruption of some 'tough' bacteria including Streptococcus mutans, Streptococcus haemolyticus, Staphylococcus aureus, and Micro-coccus lysodiekticus. Up to 5 kg per hour of such bacterial paste could be treated using the laboratory scale machine with a 600 ml continuous-flow grinding container. The effect of various parameters on breaking of yeast cells has been discussed. Kula have studied the release of several enzymes from brewers' yeast under various conditions. Rehacek studied the disintegration of yeast cells in a novel type of agitator in which the agitator discs were placed alternately. Perpendicularly and obliquely on the drive-shaft, an arrangement which was claimed to be more efficient. Recently Woodrow examined the conditions necessary for the optimum release of a number of bacterial enzymes using a Dyno-Mill fitted with a 600 ml continuous flow chamber. The Dyno-Mill type of cell disruptor has the significant advantage that it can be readily mounted in a safety cabinet when pathogenic or genetically engineered microorganisms are to be disrupted.
Liquid Shear
Milner used a liquid shear system for the disruption of chloroplasts. The suspension was passed through a needle valve at pressures up to 20000 p.s.i. (137 MPa) and a flow rate of 10 ml/min. Under these conditions, rupture of E. coli was indicated by an increase in glutamic acid decarboxylase activity. Ribi improved this procedure with an apparatus working at a loading of 40000 p.s.i. (275 MPa). The outlet temperature was controlled at 15°C with gaseous CO2, and the apparatus was capable of handling 300mg of bacterial protein.
More recently the APV Manton Gaulin homogenizer has been used for disrupting bacteria on a continuous basis. The machine is a positive-displacement pump, incorporating an adjustable valve with a restricted orifice. The cells in liquid suspension are passed through the homogenizer at pressures up to 55 MPa. There would appear to be three distinct mechanisms causing disruption of the cells. The cells are pumped through a non-return valve after which they impinge on the homogenizing valve at the selected operating pressure. Subsequent to this, they pass through a narrow channel bounded by the homogenizing valve and an impact ring. It is at this point that the greatest shear forces are exerted. The pressure is then rapidly reduced to atmospheric pressure as the suspension passes into the outlet. This sudden reduction in pressure is the third force exerted on the suspension and having a disruptive effect. Charm indicated the three ways in which the homogenizers can be used: single pass, batch recycle, and continuous recycle and bleed. They also gave equations for determining the number of whole cells remaining, and the overall processing rate of the methods. Protein release on disruption of yeast cells is described by the first order rate equation.
Log(Rm/Rm-R) = KNP2.9
R = amount of soluble protein released in g per gm of packed cells
Rm = the maximum amount of soluble protein released
K = a temperature-dependent rate constant
N = the number of times the suspension is passed through the machine
P = operating pressure
2.9 = power variation
Protein release was found to be coincident with cell disruption, although some enzymes were released more slowly, and some more quickly than the mean protein release rate. These differences were not sufficient, however, to achieve significant purification. Although such rate equations may be used to describe protein release under well-defined conditions, in practice their value is limited, and conditions need to be empirically determined.
Gram-negative bacteria are, in general, more easily disrupted then Gram-positive organisms and the history of the material including such factors as growth conditions and freezing and thawing affect the protein release rate. In addition, the rate of liberation of some enzymes may differ from that of the majority of the cell protein. In particular, periplasmic enzymes are more readily liberated and recently a very mild homogenization treatment (3.5 MPa) has been found to remove surface appendages from E. coli without disrupting the bacteria.
ISOLATION AND PURIFICATION
NUCLEIC ACID REMOVAL
In the context of this chapter nucleic acids will be considered as contaminants, and the interest is in their removal from an extract while maintaining enzyme activity.
Factors such as shear high pH, low ionic strength and the presence of nucleases lead to nucleic acid denaturation, and although in the purification of nucleic acids these conditions should be avoided in enzyme purification such factors can be of positive help.
A second group of techniques which may be more correctly considered to remove nucleic acids are those involving precipitation. They appear to act by complex formation between the negative phosphate residues of the nucleic acid molecules and positively charged groups of the precipitant. The resulting complex may then be removed by centrifugation.
Cetyltrimethyl Ammonium Bromide
The isolation of bacterial nucleic acids from Mycobacterium tuberculosis, Mycobacterium phlei, and Sarcina lutea by precipitation with the cationic detergent cetylmethyl ammonium bromide (cetavlon) has been described by Jones. Guerritore demonstrated that precipitation of a solution of RNA in water was a function of pH, salt concentration, and the nucleic acid/cetavlon ratio. Using ribonucleic acid at a concentration of 500 mg/ml in water at pH 7.0 and a temperature of 20°C, they found that the optimum nucleic acid/cetavlon ratio was 1-2. Sodium chloride, sodium sulphate, and sodium citrate all increased the solubility of the nucleic acid at concentrations above 0.2 M; while glycine, glucose, and urea had little or no effect. In these salt solutions the precipitation remained unaffected within the pH range 5.0 to 9.0.
Streptomycin Sulphate
Snoswell developed a technique for removing nucleic acids from bacterial extracts using streptomycin sulphate. This method was designed because of difficulty encountered in using other methods. Protamine sulphate and manganese chloride have been used and often proved difficult to reproduce leading to a considerable loss of protein.
The precipitation of nucleic acids is dependent on salt concentration and for this reason Snoswell found it necessary to dialyse their ammonium sulphate extracts thoroughly before adding streptomycin. They found that precipitation was dependent on pH and the ratio of streptomycin/protein. The optimum pH ranged from pH 6-8, and the best results were obtained with a ratio of streptomycin/protein of 1.0. With an extract of Lactobacillus plantarum and using a solution of 10 mg/ml protein, 10% (w/v) of streptomycin at pH 7.0, a conductivity of 0.38 mS and a streptomycin/protein ratio of 1.0, only 24% of the protein was lost. This compared favourably with other methods and was preferred because of its reproducibility.
Apart from loss of protein when using streptomycin, there are potential safety problems. Streptomycin itself is toxic, and the use of large quantities may lead to over-exposure of the operators. Also, continual use may encourage streptomycin-resistant bacteria which may cause difficulties if streptomycin needs to be used therapeutically.
Polyethyleneimine
Polyethyleneimine is a long-chain cationic polymer with a molecular weight of about 24000, and it has been reported to be highly effective for the precipitation of nucleic acids. By adding polyethyleneimine to a cell extract following breaking, it was found possible to remove precipitated nucleic acids and cell debris simultaneously by centrifugation. Using a synthetic DNA-RNA mixture with a concentration of polyethyleneimine of 0.294% in the presence of 0.02 M NaCl at pH 7.0 and a temperature of 40°C. 89% of the RNA and 96% of the DNA was precipitated. Precipitation with polyethyleneimine has also been used to selectively purify the restriction endonuclease E. coli.
With an extract of E. coli EM 20031 it was shown that the effectiveness in precipitating nucleic acids decreased in the order polylysine ] polyethyleneimine ] Cetavlon ] streptomycin sulphate ] protamine sulphate ] MnCl2. Spermine, spermidine, tetramethyrenediamine, and MnCl2 were relatively inefficient and resulted in over 75% of the nucleic acids remaining in solution. Extracts of Micrococcus lysodiekticus treated with protamine sulphate showed a loss of catalase activity and ineffective removal of RNA. In contrast, treatment of a similar extract using polyethyleneimine resulted in 90% precipitation of the nucleic acid and 70-% recovery of catalase and oxaloacetate decarboxylase.
Difficulties have been encountered using polyethyleneimine, and it was found that some enzymes of Thermus aquaticus were precipitated by polyethyleneimine, although this could be overcome by increasing the ionic strength. Triosephosphate isomerase from B. stearothermophilus has a high affinity for nucleic acids, and was co-precipitated with the nucleic acid. In addition, dihydrofolate reductase from L.casei complexed with polyethyleneimine causing enzyme inactivation. Activity could, however, be restored by extensive dailysis, during which time the polyethyleneimine was removed. Trim noted depolymerization of nucleic acids when bacterial extracts were prepared by liquid shear, and it was demonstrated that only 85% of nucleic acids were removed in this type of preparation compared with 95% when extracts were obtained by non-shear methods. Finally, one severe limitation on the use of polyethyleneimine is the fact that it may be carcinogenic owing to the presence of unreacted monomer.
Nuclease Treatment
This has become the method of choice in recent years. It is well known that bovine pancreatic ribonuclease and deoxyribonuclease can be used to hydrolyse bacterial nucleic acids, and deoxyribonuclease treatment of disrupted bacterial suspensions has been reported, although not investigated in detail.
Melling examined nuclease treatment as a method for the removal of nucleic acids from bacterial suspensions. It was clear that with the two strains of E.coli used, nuclease treatment was effective in depolymerizing nucleic acids and hence improving the recovery of supernatant after centrifugation to remove cell debris. However, at this stage there was a relatively high nucleotide content, amounting to some 15-20% of the total protein plus nucleotides. This could be reduced to a very low level by ammonium sulphate precipitation and subsequent dialysis of the precipitate. Since such steps are commonly used during an extraction procedure, it appeared to provide a useful means of reducing nucleic acid contamination.
It was apparent that an organism's own nucleases may provide in often unrecognized 'nuclease treatment', and the results with a ribonuclease-deficient strain of E.coli (MRE 600) emphasized the importance of ribonuclease as well as deoxyribonuclease treatment if removal of nucleic acids was to be effective.
The cost of nuclease treatment of 5 kg of bacterial paste was £1, compared with £30 for streptomycin sulphate and £350 for protamine sulphate.
CONCENTRATION BY PRECIPITATION
Ammonium Sulphate
Salting out of proteins has been employed for many years and has fulfilled the dual purpose of both purification and concentration of specified proteins. The salt most commonly used is ammonium sulphate, because of its high solubility, lack of toxicity to most enzymes, cheapness and in some cases, its stabilizing effect on enzymes.
The theory of enzyme fractionation by salting out has been discussed by Green. In concentrated electrolyte solutions the logarithm of the decrease in protein solubility is a linear function of increasing salt concentration (ionic strength). The general equation describing the salting out process is:
log s =
where:
s = solubility of protein in g/1 of solution
Bl = intercept constant
Kls = (slope of line) salting out constant
T = ionic strength (moles/1)
The value Bl is dependent on the salt used, but will vary with pH, temperature and the nature of the protein in solution. Kls, on the other hand, is independent of pH and temperature but will vary with the protein in solution and the salt used.
A protein solution of known concentration(s) will begin to precipitate at the ionic strength given by the following equation:
As pointed out by Charm reproducible results can only be obtained if pH. temperature, and protein concentration are kept constant, and under these conditions the salt concentration required to precipitate an enzyme will vary with the concentration of enzyme.
There are some useful points to consider when varying such parameters as pH, temperature, and type of electrolyte. At constant ionic strength the solubility of a protein increases when it is on the acid or alkaline side of its isoelectric point. This means that a reduced ionic strength will precipitate a protein if held at its isoelectric point. In dilute electrolyte solutions proteins are more soluble at temperatures above 0°C. However, in concentrated electrolyte solutions this effect may be reversed. Salts of a high valency produce higher ionic strengths than low valency salts and are therefore more efficient in the precipitation of proteins.
The use of ammonium sulphate in enzyme purification has been described by many authors. The extraction of penicillinase from E. coli strain W3310 is a fairly typical example. Ammonium sulphate (20%, w/v) precipitation of unwanted protein improved the specific activity 4-fold, and increasing the ammonium sulphate concentration to 56% (w/v) to precipitate the penicillinase resulted in a 5-fold purification overall coupled with a reduction in volume from some 60 litres to a paste weighing 2.4 kg.
Organic Solvents
The addition of organic solvents to aqueous solutions of proteins reduces the solubility of the proteins by reducing the dielectric constant of the medium. As increasing amounts of organic solvents are added, protein molecules tend to interact more with other protein molecules than with water. Complexing of oppositely charged protein molecules continues until a point is reached at which the protein is precipitated. This occurs as a result of the decrease in the dielectric constant, and hence an increase in the coulombic attractive forces between the unlike charges of protein molecules. Addition of salt to an aqueous organic solvent increases the solubility of the protein. Temperatures above 4°C can lead to denaturation in the presence of organic solvents, and for this reason it is desirable to work at temperatures below 0°C, which is feasible because of the depression of the freezing point produced by organic solvents.
As pointed out by Green the effects of organic solvents may be due to factors other than modification of the dielectric constant. It was suggested that the exchange of protein-bound water for molecules of the organic solvent may lead to solvation of the protein. Because the organic solvents used have to be water miscible it was also suggested that the protein might precipitate owing to dehydration. However, these effects are unpredictable and probably depend on the degree of ionization of the protein such that the presence of many charged groups would possibly lead to dehydration, and few to solvation.
Various organic solvents can be employed for the precipitation of proteins, with methanol, ethanol, and propan-2-ol being the most important, although acetone and even diethyl ether can be employed. These latter two solvents have the serious disadvantage of greatly increased flammability.
For large-scale work, the precipitation and concentration of enzymes with organic solvents has not been widely used. Although the degree of precision by which a protein can be precipitated using this method is high, the flammable nature of the materials together with a relatively high cost has made them less attractive than other methods, with the notable exception of serum fractionation. In this process, despite the introduction of other techniques, ethanol precipitation has reigned supreme since its introduction by Cohn. This process has been developed into a highly automated computer-controlled system.
High Molecular weight Polymers
Other organic precipitants which can be used for the fractionation of proteins are the water soluble polymers, of which polyethylene glycol is the most widely used. This has the advantages of being non-toxic, non-flammable, and non-denaturing to proteins. Its mode of action and applications have been described by Kula, but it is not widely used, except in the blood processing field.
Concentration by Ultrafiltration
Although the technique of ultrafiltration is considered here as a method for concentration, it can nevertheless constitute a valuable means of achieving a purification of some desired proteins by exploiting differences in molecular weight (size) between the required protein and other unwanted proteins.
Ultrafiltration should be considered as one part of a spectrum of filtration techniques ranging from reverse osmosis (molecular weight cut-off up to about 250), ultrafiltration (molecular weight cut-offs between 500 and 300 000), micro-particle filters up to particle filtration, with some overlap between the ultrafiltration and micro-particle ranges.
There are two types of ultrafiltration membrane, the microporous ultrafilter and the diffusive ultrafilter. The microporous ultrafilter can best be described as similar to the conventional idea of a filter. The membrane is rigid with a series of extremely small random probes running through it with an average size of the order of 500-5000 A. Thus, very small molecules will pass through the membrane whereas very large ones will be retained within the structure of the membrane. It is because these filters retain intermediate size molecules that they are susceptible to blockage. This leads to the smaller pores being blocked first, leaving only the larger pores for passage of solute and solvent. The result is that the filter becomes less retentive and does not discriminate so well between different solutes. However, the ultimate result is blockage to most molecular species except the very smallest, and thus a finer pore filter may be formed. To minimize blockage or fouling it is advisable to choose a membrane that has a mean pore size well below that of the solute to be retained. From this it can be deduced that the microporous membranes are useful for the concentration of solutes of high molecular weight ([1 X 106). Microporous filters have, however, been largely superseded by membranes of the diffusive type, bacause as well as concentration they are capable of more selective molecular discrimination.[/P]
The diffusive ultrafilter is essentially and homogeneous hydrogel membrane through which solvents and solutes are transported by molecular diffusion under the action of a concentration or activity gradient. The transportation of a molecule requires considerable kinetic energy and is a thermally activated process. Michaels considered that the energy required depends mainly on the dimensions of the diffusing molecule, the density of the membrane polymer matrix, as well as the forces of interaction between the diffusing molecule and the matrix, and the constraints upon free motion of the matrix polymer segments. Thus, for a membrane of high permeability, the conditions would be a highly hydrated gel matrix and a strong specific affinity between polymer and solvent. Conversely, for a membrane of low permeability the conditions and reduced affinity between solute and polymer.
The first recorded demonstration of ultrafiltration was by Schmidt in 1861. In this he partially retained gum arabic, using bovine pericardium as a membrane. The first artificial membrane was constructed by Martin which he used in the fractionation of snake venoms. Bechold introduced the term ultrafiltration and also the use of flat reinforced collodion membranes. A breakthrough due to the development of anisotropic diffusive membranes was made between 1961 and 1965. It is largely owing to these developments that ultra-filtration membranes are such useful tools in the isolation and purification of enzymes on a large scale.
The anisotropic membrane consists of a highly consolidated, but very thin skin (0.1-5 mm) supported by a relatively thick (20 mm-1 mm) porous substructure which acts as a support. Because the active layer is so thin, such membranes exhibit extremely high flux rates, while the porosity of the support is such that any molecule passing through the membrane, is not retained by the support. The advantage of this type of membrane. which contains no pores in the conventional sense, is that it does not 'plug' or block within the membrane, and so there is no reduction in solvent permeability at constant pressure.
Solute transport through these membranes is largely governed by Pick's law of diffusion, within the membrane. Thus, solute flux is proportional to the solute concentration gradient which is determined by solute concentrations in the 'concentrate' and 'ultrafiltrate', and is independent of pressure. This means that as solvent flux is increased by an increase in pressure the solute flux is little altered, so that the retention efficiency of the membranes increases.
Although the diffusion coefficients of various solutes differ according to the shape and size of the molecules, they tend in general to vary exponentially with the square of the molecular diameter.
At relatively low pressures the solvent activity is nearly directly proportional to hydrostatic pressure. However, as already mentioned, solvent permeability is temperature dependent. This is because temperature influences the activation energy for the diffusion of solvent within the membrane. Temperature will also affect the fluidity of the solvent, but is not significant in this process.
Although high solvent flux rates are readily obtained by using anisotropic membranes, the flow of solvent may be drastically reduced by a phenomenon known as concentration polarization. This arises as a result of the build-up of a layer of rejected solute at the membrane surface which impedes solvent flow. The resistance to flow due to this layer thus increases until a point is reached where the rate of diffusion of solute away from the polarization layer equals the rate of deposition. This phenomenon of concentration polarization also explains why the solvent flux often becomes invariant with an increasing transmembrane pressure drop. Although solvent flux through a membrane is a hydraulic pressure-activated process and should increase with increasing pressure, a point is reached where the rate of build-up of the solute polarization layer is such that flow is impeded and further increases in pressure have no effect.
The design of ultrafiltration equipment has been aimed at keeping such polarization layers to a minimum. In laboratory apparatus this has been achieved with a stirrer to increase the rate of removal of solute from the layer. In larger equipment high flow rates through hollow channels have been employed.
The ultrafiltration characteristics of proteins may vary, depending upon a number of factors. Where this does occur it was suggested that a high solute concentration should be used, presumably to saturate the membrane sites without affecting the recovery too significantly. Melling studied the ultrafiltration characteristics of a partially purified preparation of penicillinase derived from E. coli. Using a 30 000 molecular weight cut-off (anisotropic) membrane the throughput of enzyme varied from 1.6% to 55% depending upon buffer pH and salt concentration. Maximum throughput was at pH 5.1-6.2 which is near the isoelectric point of the enzyme, where penicillinase-protein interactions might be expected to be minimal. At pH 8.0 the throughput could be increased from 1.6% to 20% by increasing the buffer concentration from 1 to 30 mM and was possibly due to some buffer-shielding effect on penicillinase-protein interactions. The addition of EDTA also increased enzyme throughput, while urea had the reverse effect.
Salt concentration, the nature of the salt used, and pH all affected the retention of a b-lactamases from Staph aureus, B. cereus, and E. coli as well as a b-N-acetyllglucosaminidase from B. subtilis. The case of b-N-acetylglucosaminidase is of particular interest since it was shown that the pattern of liberation of this enzyme from the bacterial cell with increasing NaCl concentration was reflected by the effect of NaCl on enzyme retention by an ultrafiltration membrane. As well as effects related to pH and salt concentration, specific protein-protein interactions may affect the retention of a protein. Elongation factor Tu has a throughput of 45% with a 100 000 molecular weight cut-off membrane, but addition of factor Ts, which is known to form a complex with Tu reduced throughput to 8% (A. Atkinson, unpublished results).
By manipulating those factors which influence the ultrafiltration characteristics of proteins, considerable increases in purification may be obtained, for example 7-fold for E. coli penicillinase. In view of this, and taking into account the relative ease of scale-up from laboratory processes, ultrafiltration should provide a powerful technique for large-scale work in enzyme purification as well as concentration. Besides its unquestioned role as a means of concentration, surveys of enzyme purification suggest that ultrafiltration for concentration is used in the majority of protein purifications. Ultrafiltration can also be used for the rapid removal of low molecular mass solutes, such as ethanol or water from protein solutions, a process which has been termed diafiltration. In this process the liquid lost as ultrafiltrate is continuously replaced by fresh buffer, thus maintaining a constant volume of solution in the ultrafiltration cell. It can be calculated that the removal of 99% of the solutes requires the throughput of 4.6 X the initial solution volume. If necessary the throughput volume can be reduced by an initial concentration step. By suitable choice of ultrafiltration apparatus this technique is applicable to any scale of operation ranging from a few millilitres to hundreds (or thousands) of litres. Finally, it is worth noting some of the characteristics which make ultrafiltration a useful method in enzyme work.
- A low temperature operation, if desired thus, by ultrafiltration at a cold room temperature, -5°C to +5°C, loss in activity due to exposure at high temperature is avoided and autodigestion can be minimized.
- No phase change required.
- Operation at low hydrostatic pressure.
- Gentle and non-destructive.
- No chemical reagents required.
- Simultaneous concentration and purification if desired.
- Maintenance of constant ionic strength and pH of the concentrate avoiding inactivation of the enzyme.
- Economical.
Concentration by Freeze-Drying
A number of authors have reviewed the process of freeze-drying and accordingly no detailed discussion of the basic process will be attempted here.
Although, in certain instances, freeze-drying may be employed for the concentration of proteins, its use is limited by the fact that unless the salt concentration in solutions is sufficiently reduced, eutectic mixtures may be formed. This may lead to incomplete drying or severe foaming and protein denaturation. Such problems can be avoided by careful control of the temperature of the frozen mass to maintain a temperature below that of the lowest partial eutectic. It may also be necessary to reduce the salt concentration before commencing the freeze-drying process.
In view of the other methods of concentration which are now available for protein solutions, freeze-drying may best be reserved for the preparation of materials for storage or transportation.
Many different proteins have been freeze-dried without denaturation or loss of activity. However, it is not uncommon to find that some proteins show denaturation and complete loss of biological activity. This appears to be especially true for these proteins which are composed of subunits, and the studies of catalase and myosin provide useful information in this respect. Rosenkranz found, by examination of ORD and CD spectra, that the structure of E. coli asparaginase was significantly increased by freeze-drying. Marlborough found that freeze-drying (as opposed to freezing and thawing) of Erwinia carotovora asparaginase appears to perturb the tertiary and quaternary structure of the enzyme molecule, with the consequent weakening of the interaction between subunits. However, if the freeze-dried product was reconstituted in buffer solution within the pH range 5.0 to 7.0 these perturbations were reversed. Using buffers with pH values outside these limits resulted in loss of activity and denaturation.
In the light of these observations, the freeze-drying of enzymes should not only be carefully controlled, but it is also clear that it may be necessary to investigate the process in some detail for each enzyme. In particular where materials are for clinical use these requirements are especially important.
Gel Chromatography
In this section, as in the others, it is the preparative, rather than the analytical, applications of the technique which will be emphasized. The interest in this technique is based on its ability to fractionate and purify enzymes. From this viewpoint, consideration will be given to the gross structure of gels, and to the general theory which accounts for the separation of biologically active compounds, according to their molecular size and shape.
As pointed out by Ackers, confusion has arisen owing to the number of titles applied to this subject, and it might therefore be of use if these were listed:
- Gel filtration.
- Molecular sieve filtration.
- Molecular sieve chromatography.
- Exclusion chromatography.
- Restricted diffusion chromatography.
- Gel permeation chromatography.
- Gel chromatography.
- Steric chromatography.
For simplicity and adequacy, Ackers preferred the title 'Gel chromatography' to describe this subject, and this will be used throughout this section. Should readers wish to review the field in detail, they are referred to Ackers where a number of useful references can be found.
The observations by Wheaton suggested the principle of gel chromatography. In 1959 Porath & Flodin demonstrated the separation of proteins by using crosslinked dextrans in a chromatographic process. Commercial pressure then led to development of the Sephadex (Pharmacia, Uppsala, Sweden) series of gels as well as several other products.
The principles of gel chromatography can be illustrated by reference to the Sephadex gels, crosslinked with epichlorohydrin. The dextran used is a soluble polysaccharide of glucose and is a product of the microorganism Leuconostoc mesenteroides. The glucose residues are predominantly a- 1- 6 linked glucosidic bonds, the dextran chains being crosslinked via glucose hydroxyl groups, resulting in glycerin ether bonds. The degree of crosslinking is controlled by the amount of epichlorohydrin used, which in turn controls the degree of hydration (water regain) within the gel matrix and hence the porosity. The greater the water regain per unit of dry weight of gel, the larger the molecular species which can be fractionated. Conversely, an increase in the amount of dextran matrix present per unit dry weight of gel, leads to a reduction in the size of the molecules which can be fractionated. Thus, if a mixture of high and low molecular weight material is placed on top of a gel column of low water regain (that is, highly crosslinked), the high molecular weight species will pass down the column in the mobile phase, and will be collected first. The low molecular weight species will be retarded owing to its passage through the stationary phase, which is inside the gel.
The process of gel chromatography is the diffusional partitioning of solute molecules between the readily mobile solvent phase and that confined in spaces within the porous particles that make up the stationary phase. The mechanism of gel chromatography may be more easily understood by considering the gel as having a porous bead-like structure. The mobile phase is that liquid external to the gel particles, known as the void volume (Vo), while the stationary phase is that liquid which occupies the interior of the gel particles (Vi). Diffusional exchange of solute takes place between the stationary and mobile phases. The only volume unaccounted for in the total volume of the column (Vt) is that occupied by the gel matrix (Vg). Therefore:
Vt = Vo + Vi + Vg
Taking Sephadex G75 as an example, the fractionating range of this material is from 3000 to 80 000 MW and will, to varying degrees, be affected by diffusional partitioning between Vo and Vi. Molecules within this range will be fractionated according to their molecular weight, shape, and size, giving each molecular species its own partition coefficient. This parameter represents the extent to which a given molecule penetrates the stationary phase. Mathematical calculations for partition coefficients have been given by Ackers, but simply, the partition coefficient Kav can be obtained from the following equation:
Kav = (Ve - Vo)/(Vt - Vo)
Ve = volume of solvent required to elute solute from column
Vo = void volume
Vt = total volume of column bed.
The value of Kav has been shown, empirically, to be inversely proportional to the log of the molecular weight. Figure 1 illustrates the relationship for globular proteins on Sephadex G75. From this curve it can be seen that three variants of gel chromatography are possible; A to B desalting, B to C molecular fractionation, and C to D exclusion.
We have briefly explained the mechanism of gel chromatography. However, it should be noted that there are four basic theories governing gel chromatography, (1) The Exclusion Theory, (2) The Theory of Restricted Diffusion, (3) Partition due to Molecular Surface Forces, and (4) Partition due to the Osmotic Pressure in the Gel. As yet a unifying theory has not appeared.
In recent years a variety of materials other than crosslinked dextrans have been introduced as matrices for gel chromatography. These include polyacrylamide, agarose, polystyrene, crosslinked agarose, agarose-polyacrylamide mixtures. porous glass, and polymethylmethacrylate. Most of these materials offer significant advantages over the conventional gels, particularly when the scale-up of a chromatographic separation is contemplated. In particular, the particles are considerably more rigid, smaller, and uniform in size, resulting in improved resolution and flow properties.
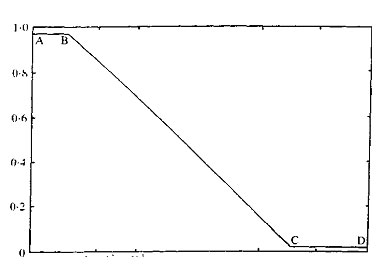
Fig. 1. Relationship between Kav and molecular weight for globular proteins
Ion Exchange Chromatography
There are three types of ion exchanger available: the ion exchange resins, ion exchange celluloses, and ion exchange dextran and agarose gels. The latter types have found the widest application, but the ion exchange resins have proved valuable on a more limited scale.
Ion exchange resins
These may be generally described as water insoluble polymers containing either anionic or cationic groups. The nature of the functional group varies, but they are of the general form: cationic exchangers RH+ and anionic exchangers ROH where R represents the resin polymer. Exchange of either H+ or OH ions will take place for ions combined with some weaker acid or base, in this case the proteins, and thus the protein molecules become re-versibly attached to the resin.
The functional groups found in cationic exchangers include -SO3H,-COOH, and -OH in decreasing order of acidity, while anionic exchangers often contain quaternary ammonium groups -NH+3Cl (or other anions).
Resins have the advantages of being stable to the physical stresses usually encountered, with a high capacity for protein adsorption, they sediment rapidly, and when packed into columns allow high liquid flow rates. The high capacity of these materials (up to 5 meq/g) is often disadvantageous when considering their use for protein purification, since the protein may not withstand the harsh conditions necessary for elution from the resins.
lon exchange resins have nevertheless been used successfully in some protein purifications
Ion echange celluloses.
A variety of charged groups, either cationic or anionic, may be introduced into the cellulose by various chemical processes However, the level of substitution is usually very low, and the capacity of ion exchange celluloses is about 1/10th that of resisns. This allows the elution of polyelectrolytes under mild conditions. In addition, substitution above 1 meq/g may lead to solubilization of the cellulose, which is clearly undesirable. Table 1 indicates some of the types of celluloses which are available, together with their properties.
Other ion exchange gels.
Cellulose has been the traditional medium for the ion exchange chromatography of proteins. In recent years, however, a number of other ion exchange materials have been introduced, which generally have superior flow properties. Ion exchange Sephadex gels are prepared by introducing DEAE on CM groups into Sephadex G25 or G50 (Pharmacia). Those based on Sephadex G50 have a high capacity for proteins but unfortunately are rather soft and tend to shrink and swell when the pH or ionic strength of the buffer is varied.
Table 1
Characteristics of Various Cellulosic Ion Exchangers

The under alkaline conditions. of DEAE or CM groups into crosslinked agarose or acrylic copolymers gives ion exchange Sepharoses (Pharmacia) and Trisacryl (LKB). These two types of exchangers have a high capacity for proteins, and small. uniform bead shaped particles which support high flow rates. They do not deform under the pressures normally encountered in chromatography. and more importantly do not shrink or swell when the pH or ionic strength of the buffer is changed. As they do not even shrink or swell at extremes of pH they can be regenerated in the column, a considerable advantage in large-scale operations. One further advantage of these types of exchanger is that they can be sterilized repeatedly when they are being used for the purification of therapeutic products.
The one major disadvantage of these materials is their high cost, particularly when compared with the more traditional materials like cellulose. However, in many cases the higher flow rates obtainable means that the extra cost is recoverable by a greater production capacity.
Ion exchange chromatography can differentiate between components in a polyelectrolyte mixture on the basis of the charge carried by the individual polyelectrolytes. However, the net charge, charge density, and molecular size of the protein as well as the pH and ionic strength of the solution are all parameters which affect this chromatographic process. The partition coefficient is the expression which relates the number of molecules of a particular electrolyte in the solvent to the number attached to the stationary phase at equilibrium. Therefore, the greater the number of equilibrations a polyelectrolyte species is subjected to the greater is its resolution. Efficiency of separation is also related to an exchanger's capacity; the greater the capacity the larger the number of equilibration, which in turn reduces the differences in partition coefficient required to separate different electrolytes.
In the large-scale isolation of enzymes, however, column chromatographic are usually reserved for the final stages of purification, owing to the large volumes which may have to be handled at the beginning. However, ion exchangers can be of considerable use in the early stages of enzyme isolation when employed in a 'batch' technique. This involves binding of the polyelectrolyte to the exchanger such that at any given time the greater part of the polyelectrolyte is associated with the matrix. Thus it can be said that the polyelectrolyte is immo- bilized. The conditions generally required for the immobilization to occur are obtained when the electrostatic bonds formed are strong. This requires that the charge on the polyelectrolyte is high and opposite to that of the exchanger. Normally, basic polyelectrolytes (isoelectric point above pH 7.0) are adsorbed onto cationic exchangers (for example, CM cellulose) at a pH below the isoelectric point, and acidic polyelectrolytes (isoelectric point below pH 7.0) onto anionic exchangers (for example DEAE cellulose) at a pH above the isoelectric point. Therefore, it is important to know the isoelectric point of the particular polyelectrolyte. After choosing the exchanger to be used, the conditions of pH and ionic strength are of prime importance. The pH has to be of sufficient extreme to give a high charge on both exchanger and polyelectrolyte; while the ionic strength should be chosen such that the salt helps to preserve the activity and solubility of the enzyme, but is not high enough to compete effectively with the enzyme for sites on the exchanger. If the conditions are chosen correctly it is possible to adsorb the enzyme onto the exchanger, wash with buffer to free it of all non-bound proteins, and then elute the enzyme from the exchanger.
The eluting methods in common use are change of pH, increase in ionic strength, of a combination of both. By changing the pH of a suspension of a cationic exchanger, with an adsorbed basic enzyme, from acid to alkaline the charge of both the exchanger and the enzyme is reduced, thus the electrostatic bonds are broken and the enzyme released. When the ionic strength is increased, competition for the sites occupied by the enzyme is increased, until a point is reached when the enzyme is again released. A combination of both processes is useful when the enzyme is particularly sensitive to changes in ionic strength and pH. Fractional desorption can be advantageous when several polyelectrolytes are adsorbed to the same exchanger. This can often be controlled by varying both pH and ionic strength. Enzymes whose isoelectric points are well removed from neutral are generally more easily purified by using ion echange chromato-graphy by the 'batch' method. With L-asparaginase which has an isoelectric point of 8.6, a 100-fold purification is possible using CM cellulose batch-wise.
The batch technique is also of use in removing contaminating polyelectrolytes such as nucleic acids. The increase in protein purification may be marginal, but later steps are often aided by the removal of these large, highly charged polyelectrolytes.
Batch adsorption and subsequent elution may appear to be crude and not to exploit the fractionating ability of the ion exchange technique to the full; however, it has proved a most useful technique which allows simultaneous purification and concentration to be achieved prior to the application of more sophisticated methods.
Affinity Chromatography
Affinity chromatography can provide perhaps the most elegant solution to the problem of purifying individual proteins from a complex mixture, However, affinity chromatography, although frequently employed on a laboratory scale, is only rarely employed for the large-scale purification of proteins.
Affinity chromatography relies on the more or less specific interaction of a protein with an immobilized ligand. Such ligands fall into the two main groups:
- Those which are specific for the desired protein, in which case the ligand is generally an antibody, a substrate, or substrate analogue or an inhibitor.
- Those which can interact with a variety of proteins. Such genera ligands may be specific for different classes of enzymes such as 5' AMP. 2',5' ADP, or NAD+, or they interact with a wide range of proteins such as hydrocarbon chains or dyes.
Whatever the nature of the interaction it is essential that it be reversible. A wide variety of eluants can be employed to desorb a protein from an affinity ligand. ranging from nonspecific methods such as an increase in ionic strength or alteration in pH to specific methods using a cofactor or free ligand.
The matrix to which the ligand is attached should exhibit the following basic properties:
- Weak reactivity with proteins in general, to avoid nonspecific adsorptions.
- Show good solvent flow rates after coupling of ligand.
- Contain chemical groups that can be modified such that the matrix is not damaged.
- These chemical groups should be plentiful, leading to a high capacity after coupling.
- Must be mechanically and chemically stable under the various conditions required for coupling and for elution.
- Have a loose porous and hydrophilic network allowing free passage to large macromolecules, preferably be uniform, spherical, and rigid.
A large number of studies on the effects of different matrices on affinity chromatography have been carried out. To date the most frequently used matrix is 4% agarose in bead form, which satisfies most of the desired criteria and is readily available.
Coupling techniques
The most frequently employed coupling procedure employs cyanogen bromide under alkaline conditions to generate a reactive imidocarbonate which can be subsequently reacted with a nucleophilic moiety on the ligand. Detailed studies on the chemistry and quantitation of the cyanogen bromide activation process have been carried out by Kohn.
Bisoxiranes (bisepoxides) such as l,4-bis-(2,3-epoxypropoxy-) butane form ether linkages with a variable length hydrophilic spacer between the matrix and ligand. They can be used to couple ligands containing hydroxyl, amino, or thiol groups. Other coupling reagents have been proposed such as toulene-p sulphonyl chloride (tosyl chloride) or 2,2,2 trifluoro ethane sulphonyl chloride (tresyl chloride), which from stable linkages between the matrix and amine-containing ligands. Carbonylating reagents such as l,l'-carbonyl di-imidazole or l , l'-carbonyldi-l,2,4-triazole can also be used for the rapid coupling of labile, amine containing ligands.
Despite the introduction of numerous other coupling reagents, the cyanogen bromide method remains the most widely used, despite the toxicity of cyanogen bromide, because it is relatively simple and gives good coupling yields. The cyanogen bromide technique can also be used to introduce hydrophobic spacer groups into the matrix by coupling 1.6-diamino hexane or 6-amino hexanoic acid to a cyanogen bromide activated matrix.
These coupling techniques can be used to couple small molecules such as cofactors or substrates, or large molecules such as antibodies to a solid support.
In general terms absorption takes place under conditions of low ionic strength and neutral pH Nonspecific elution can be effected by change in pH, ionic strength, or temperature. Biospecific elution, which will often give a higher degree of purification, can be effected by substrate or substrate analogue.
Despite the vast amount of literature relating to affinity chromatography and its undoubted impact on enzyme purification, reports of its use in large-scale purification are rare. The main reasons for this lack of acceptance are perhaps the expense of the matrix, the instability of many biological ligands. the complex chemistry involved in coupling them to a matrix, and the low capacity of many such ligands. However, under certain circumstances affinity chromatography can give excellent results, and Robinson have described the large-scale automated purification of b-galactosidase from E. coli using immobilised p-aminophenyl-b-galacto-pyranoside as the ligand.
One type of affinity chromatography which does not suffer from the disadvantages listed above is dye affinity chromatography using immobilized reactive dyes. Dye affinity chromatography has recently been employed for the large-scale purification of enzymes as diverse as alcohol dehydrogenase from horse liver protein kinase from pig's liver and glycerokinase from Bacillus stearothermophilus.
The dyes act as pseudo-affinity ligands and can bind a wide range of proteins, often with a surprising degree of specificity. Bound proteins can be eluted non-specifically by increasing the ionic strength or biospecifically by using a substrate or cofactor.
From a practical point of view the reactive dyes offer numerous advantages. They are readily available in large quantities, are inexpensive, and can be coupled to a support under mildly alkaline conditions without the use of toxic chemicals. Once coupled, the resulting bond is stable and the capacity of the conjugate is high. Indeed the capacity of a dye ligand can be some 20 times greater than for a nucleotide ligand in the case of bacterial malate dehydrogenase.
NON-SPECIFIC ADSORBENTS
Hydroxyapatite
Hydroxyapatite (HA) has been used in batch techniques for enzyme purification over a considerable period and column chromatography using HA was developed by Swingle. The method has only slowly been accepted by protein chemists, and the following reasons for its slow acceptance were given by Bernardi.
- Laborious preparation procedure of HA.
- The unknown mechanism of interaction of proteins with HA.
- The introduction of cellulose ion exchangers in 1956 by Peterson and Sober.
Laboratory-scale preparations of crystalline hydroxyapatite have been described by several authors. Atkinson published a cheap and simple method for large-scale production of HA which has a uniform crystal size, showed good flow rates and protein binding capacity. Commercially available HA was criticized by these latter authors as being expensive and often unreliable in terms of its crystal structure, flow rate, and binding capacity.
A mechanism of interaction between proteins and HA has been described by Bernardi including the influence of the secondary and tertiary structure of proteins in relation to their chromatographic behaviour. Parameters determining resolution in HA columns, and the theoretical basis of chromatography of structurally rigid macromolecules, have been dealt with by Kawasaki considered that chromatography on HA should be used as an additional tool in the purification of proteins because the basis of separation is different from that of either ion exchange or gel filtration.
The preparation of hydroxyapatite is based on the conversion of secondary calcium phosphate (brushite) by boiling under alkaline conditions.

The mechanism by which HA separates macromolecules is not fully understood. However, it is considered that exposed Ca2+ and PO34 sites on the surface of the HA crystals contribute to the fractionation of macromolecules. Acidic and neutral proteins are thought to bind to Ca2+ sites on HA: neither the binding nor the elution would appear to be affected by the presence of high concentrations of NaCl, KC1, or CaCl2. The elution of acidic and neutral proteins is generally with low concentration phosphate buffers (30-120 mM) at about pH 6.8. Basic proteins, however, bind to the PO34 groups on the HA crystals, adsorption and elution being strongly affected by the presence of NaCl, KCl, and CaCl2. Elution can generally be brought about by these salts and phosphate buffers ranging in concentration from 120 to 230 mM. There are a number of exceptions to these general rules. Phospho-proteins normally require high concentrations of phosphate for elution, while some basic proteins are not eluted by CaCl2, even at very high concentration. Bernardi also indicated that basic proteins bind strongly at pH 6.8. whereas basic amino acids bind test at pH 7.5. Atkinson pointed out the unpredictable elution properties of acidic proteins capable of binding phosphate groups, the example being tryptophanyl-tRNA synthetases obtained from B. stearothermophilus and E. coli. These acidic proteins, containing no phosphate groups, can be absorbed onto HA in 1.0 M NaCl and yet require 400 mM phosphate at pH 6.8, comparable to that required for very basic proteins, for elution. It was also noted that enzyme-bound cofactors changed the eluting pattern of dihydrofolate reductase on HA.
Using HA on the large scale for the isolation and purification of enzymes can give good results, indicating that it is useful to the protein chemist. Using a-16 litre column of HA loaded with 500 g of protein extracted from B. stearothermophilus, six enzymes were successfully separated: triose phosphate isomerase, glyceraldehyde-3-phosphate dehydrogenase, NADH dehydrogenase, valyl-, methionyl- and tryptophanyl-t-RNA synthetases. Methyionyl-t-RNA from 20 kg of E. coli EM 20031 containing 3.6 X 106 units was also fractionated on HA. The yield was 85%, and a 12-fold increase in purification was obtained. Valyl- and tyrosyl-tRNA synthetases, rhodanese, and glyceraldehyde-3-phosphate dehydrogenase from B. stearothermophilus could also be separated by HA chromatography.
Celite
Celite is a diatomaceous earth composed mainly of calcium carbonate with some silicate also present. This material has been used successfully in the purification of two B-lactamases from B. cereus. The enzymes were adsorbed directly from the culture supernatant at pH 7.0 and eluted at the same pH with a high ionic strength tris-zinc-citrate-NaCl buffer. Although both celite and powdered glass had been used previously only the B-lactamase I was recovered by these workers, possibly owing to the low pH used for adsorption (pH 4.5) or the higher pH for elution (pH8.5).
Whether or not celite is applicable to other enzymes is not clear, but the 20-fold purification obtained with the penicillinases suggests that such a cheap and readily available material may bear further investigation.
Hydrophobic Interaction Chromatography
Hydrophobic chromatography was developed following the observation that certain proteins were unexpectedly retained on affinity gels containing hydrophobic spacer arms. This concept was extended, and families of hydrophobic adsorbents prepared using a homologous series of hydrocarbon chains of different lengths. In practice most proteins can be satisfactorily purified using C-8 or phemyl substituents.
Hydrophobic interactions are strongest at high ionic strength, so adsorption to a hydrophobic matrix can be conveniently performed after a salt precipitation or ion exchange chromatography step. Elution can be effected by altering the solvent ionic strength, pH, or composition or by the use of a dielectric constant modifier such as ethanediol.
The effectiveness of different salts in promoting hydrophobic interaction varies, and both anions and cations can be arranged in a soluphobic series depending on whether they promote hydrophobic interactions (salting-out effect) or disrupt the structure of water (chaotropic effect) and lead to a weakening of hydrophobic interactions.
Table 2
Effect of ions on hydrophobic interactions

High Performance Liquid Chromatography
One of the most significant advanced in chromatography in recent years has been the introduction of high performance liquid chromatography. Originally intended for the separation of small organic molecules that were soluble in non-aqueous solvents, the technique rapidly developed into a form, suitable for the separation of larger, biological compounds soluble in aqueous solvents. In recent years, following the development of macroporous supports, the technique has become applicable to molecules as large as proteins and nucleic acids.
The high efficiency of HPLC columns is derived from the small particle size, with diameters in the range 5-50 (mm) one-tenth the diameter of classical soft gels. This small particle size necessitates high pressures in order to give usable flow rates. Thus the particles must be extremely rigid. Originally this entailed the use of silica, but the surface of silica particles is detrimental to proteins. However, silica can be derivatized by reaction with monochloro or monoalkoxysilanes to give hydrophilic surfaces which are more attractive to proteins. The chemistry of surface modified silica supports has been extensively reviewed in recent years.
A major disadvantage of silica is that it is unstable above pH 7-8; this has led to the development of rigid, polymeric supports which exhibit a much greater range of pH stability (for example Mono Q, Pharmacia; TSK-PW, Toyo Soda Co. Ltd) which have proved useful for high performance ion exchange chromatography in particular.
All of the normal chromatographic techniques, gel filtration, ion exchange, hydrophobic and affinity chromatography are available in HPLC. However, most of the applications of HPLC to protein purification described so far have been, confined to the small scale. Thus mg quantities of angiotensins were separated and purified by high performance ion exchange chromatography and even ng amounts of luteinizing hormone releasing hormone from milk by reverse phase chromatography. However, mg quantities of brain hexokinase have been isolated by high performance ion exchange chromatography, and lactate dehydrogenase has been purified by high performance liquid affinity chromatography, again on the mg scale. HPLC has considerable potential for scale-up because in addition to the use of larger columns, the technique can be readily automated, so as to allow the repeated injection of sample onto the same column.
ELECTROPHORETIC TECHNIQUES
Electrophoresis
In recent years the basic concepts and theories of electrophoresis have been reassessed.
The principles of electrophoresis are the same irrespective of the particular technique used, and can be simply stated as the acceleration of charged particles in an electric field; this is opposed by friction due to passage through the surrounding medium such that the particles move at a constant speed proportional to their charge. Cations move towards the cathode, and anions towards the anode. The passage of current through a solution is described by Ohm's law: V = R X I where V = voltage, R = resistance, and I = current, and in practice an increase in current can be effected by increasing the voltage. Since the ions carry the current, migration will be increased with increase in current.
The rate of movement of an ion is expressed as Q Ã- N where Q = field strength and N = the net charge on the ion. The rate of movement is opposed by the frictional force encountered by the ions as they pass through the solution. Stoke's Law states that spherical particles of radius a, moving at velocity V in a medium with viscosity coefficient h, will be subjected to a frictional force F, such that:
F = 6 ph aV
It can be seen that F will vary with changes in h which can be caused by a change in temperature. Thus the rate of movement of an ion will vary with temperature as well as applied current, molecular size, and shape. This does not imply, however, that molecules showing similar rates of migration are identical.
When an electrical current is passed through a volume element of unit dimensions, heat is generated according to the general formula:
W = heat generated
x = specific conductance
E = field of strength
A = mechanical heat equivalent.
The heat produced (which is dependent on the voltage applied) can lead to a number of undesirable effects in preparative as well as analytical techniques.
- Changes in density leading to convection currents.
- Decrease in viscosity causing increased diffusion, thus affecting resolving power.
- Increase in conductivity; however, this can be overcome by using a constant current source.
- Evaporation of volatile components leading to changes in pH, ionic strength, and conductivity. Evaporation can also cause water flow from the electrode vessels, resulting in accumulation of buffer salts near the centre of the electric field (only evident when using certain techniques).
- Denaturation of thermolabile enzymes.
Another problem is electro-osmosis. This is the transport of water from one electrode to another. It results from the support medium being charged. At neutral and alkaline pH the charge is usually negative, the charged medium therefore attempts to move towards the anode. To balance this, positively charged water molecules (H3O+) move to the cathode. This movement of water does not affect the fractionating capacity, but can cause loss of water from reservoirs, thus altering the homogeneity of the electrophoretic field.
In general terms, high ionic strength buffers result in a decrease in mobilitycompared to those of low ionic strength. The former, however, generally lead to more discrete bands or zones. pH has a dramatic effect on the mobility of enzymes in an electric field. Proteins are zwitterions, and as such can normally exist in three ionic forms. The form is dependent on the isoelectric point (pI) of the protein and the pH of the solution. A protein at its pI will be electrically neutral (NH3+RCOO-) and will not migrate in an electric field. On the acid side of its pI the species will be positively charged (NH3+RCOOH) and in alkaline solution will be negatively charged (NH2RCOO-).
The number of electrophoretic techniques available to the protein chemist for large-scale work are few. In most cases they are avoided because of equipment cost, time involved in fractionation, and that only exceptionally do they result in an improved purification over alternative methods. Of the preparative techniques available only one or two constitute truly large-scale methods.
There are several preparative electrophoretic techniques available, some of which will be mentioned. Using column polyacrylamide gel electrophoresis. up to 10 g of protein can be fractionated, while continuous curtain electrophoresis permits the application of 15 - 20 mg of protein per hour. Forced flow electrophoresis has been used in the purification and concentration of various veterinary vaccines of bacterial origin. with a projected capacity of approximately 9 litre/h. Continuous zone electrophoresis was introduced by Grassman, and a recent modification of the Philpot continuous preparative free electrophoresis technique has been introduced by Thompson. This apparatus has a flow rate of 0.5-1.5 litre/h (50 mg/ml) over a working day, enzyme recoveries varying between 80 and 100%.
Isoelectric focusing
Isoelectric focusing in beds of granulated gel-Sephadex or agarose is an attractive proposition, but the heat generated when using thicker gels limits the protein loading to about 1 g, which is not sufficient for truly large-scale operation.
Multimembrane Electrodecantation
Although now rarely used in practice, the phenomenon of electrodecantation was observed some years ago by Pauli applied the technique to separation of proteins. Potson developed the multimembrane electrodecantation apparatus which can be used for continuous or batch fractionation of protein mixtures.
The method involves placing a protein solution into a cell containing a number of semipermeable membranes. The pH of the solution is adjusted to the isoelectric point of the protein to be purified. A current is passed through the solution by two sloping electrodes. Because the required protein is at its isoelectric point it remains stationary in the electric field; all other proteins, being charged, will move either towards the cathode or the anode. This movement will cease when the migrating proteins meet the semipermeable membranes. The accumulation of protein at the membrane surface will cause an increase in density, thus it will move to the bottom of the cell. To help this decantation process a temperature gradient is created by the sloping electrodes, which means that the viscosity of the buffer is greater at the bottom of the cell than at the top. Thus the unwanted protein accumulated at the base of the cell and can be drawn off, leaving the required component in the cell. Fresh buffer can be added to fill the cell, and the process can be repeated to give further purification. In the continuous process, a series of such cells are linked together which reduces the carry-over of immobile protein in the outlet stream.
Using this technique, tetanus toxin, diptheria toxoid and human serum globulin have been fractionated. Mathies obtained a 3-fold purification of alkaline phosphatase; trypsin and deoxyribonuclease have been fractionated by Potson. Considerable loss of enzyme activity was observed in each case. Fleetwood obtained antibody and globulin with an 80% recovery and a 650-fold increase in purification. Brummelhuis have prepared antilymphocyte globulin (ALG), an immunosuppressive agent, from horse antihuman lymphocyte sera. Winchester used this technique in the purification of bovine 1-D glucosidase, acetylglucosaminidase, and a bacterial protease. The first two showed an approximate 10-fold purification, and the protease proved to be homogeneous by polyacrylamide gel electrophoresis. They also quote a throughput of 0.5-1.0 litre of 2% (w/v) of crude enzyme protein per hour, the only limitation being blockage of the membranes after an unspecified period of continuous running.
Multimembrane electrodecantation appears to be a suitable method for the large-scale purification of enzymes. To date, the method has been used by only relatively few people. However, as information becomes available and the expertise required is more easily obtained, the method may, in the future, become more generally accepted in the large-scale fractionation of enzymes.
CHROMATOFOCUSING
Although employed as a chromatographic technique, chromatofocusing is in effect a form of isoelectric focusing carried out on a bed of specialized ion exchange resin eluted with amphoteric buffers.
Chromatofocusing can combine the resolving power of isoelectric focusing with the convenience of chromatography, thus it is far more suitable to large-scale applications, although all the applications described so far are only small-scale. Chromatofocusing has been used for the fractionation of hexokinases from yeast and in the presence of 6M urea for the fractionation of bovine eye-lens crystalin.
AQUEOUS TWO-PHASE SEPARATION
Aqueous two-phase systems can be created by mixing solutions of polyethylene glycol and dextran or polyethylene glycol and certain salts, especially ammonium sulphate or potassium phosphate. Proteins and cell debris will partition between the two phases. The exact location of a particular protein depends on such parameters as its molecular weight, the concentrations and molecular weights of the polymers, the temperature, pH and ionic strength of the mixture, and the presence of polyvalent salts. Unfortunately the optimum conditions for favourable partition have to be found empirically for each protein. Two-phase systems can be effectively applied to remove debris from cell homogenates and at the same time achieve a certain degree of purification.
The two phases can in some cases be separated in a settling tank, but a more rapid and efficient separation can be achieved by using a centrifugal separation. As it is often easier to separate ligands of different density than solids from liquids, particularly on a large scale, it is claimed that this technique can be used to great advantage in large-scale enzyme preparations. However, despite these apparent advantages aqueous phase partition is only rarely used, perhaps because of the non-recoverable cost of the dextran and polyethylene glycol employed, although a recent report suggests that it is possible to use crude dextran with a considerable saving in cost.
Two-phase separation has been used for the separation and large-scale purification of pullulan-6-glucan hydrolase and 1,4-a-glucan phosphorylase from 5 kg Klebsiella pneumoniae cell paste, and RNA polymerase and glutamine synthetase from E. coli.
By coupling a ligand to polyethylene glycol it is possible to increase the partition coefficient of a protein in favour of the polyethylene glycol phase. Thus by using the dye Cibacron blue it was possible to achieve a 58-fold purification of yeast phosphofructokinase in two steps
